Cwiczenie 6
Modelowanie molekularne. Analiza budowy
przestrzennej bialka (papainy) przy uzyciu grafiki molekularnej
CEL CWICZENIA
1) Zapoznanie sie z
mozliwosciami jakie daje wspolczesna technika komputerowa (wysokiej klasy
sprzet komputerowy, grafika komputerowa, oprogramowanie) na przykladzie
programu ‘O’.
2) Zapoznanie sie z budowa
PAPAINY.
PAPAINA to bialko enzymatyczne hydrolizujace wiazania peptydowe (tj.
rozcinajace inne bialka). Bialko to zostalo wyizolowane z mleczka kauczukowego
owocow papai, a jego strukture rozwiazano w 1968 roku (J.K. Jansonius, J.
Drenth) z bardzo wysoka rozdzielczoscia 1.65 Angstrema. Lancuch PAPAINY zbudowany
jest z 212 reszt aminokwasowych i tworzy dwie domeny (pierwsza to reszty 1-18 i
112-207, druga to pozostala czesc lancucha aminokwasowego), miedzy ktorymi
znajduje sie miejsce aktywne. Reszty aminokwasowe wchodzace w sklad triady
katalitycznej to Cys25, His159 i Asn175.
Wspolrzedne atomow
PAPAINY, ktore sa potrzebne do wykonania cwiczenia, pochodza z Banku Danych
Bialkowych (Protein Data Bank, PDB).
Jest to komputerowe archiwum makromolekul. Dane z PDB zawieraja bibliografie,
sekwencje aminokwasowa, informacje o strukturze drugorzedowej oraz, przede
wszystkim, wspolrzedne atomow.
Struktura
drugorzedowa opisuje wzajemne przestrzenne ulozenie reszt aminokwasowych,
sasiadujacych ze soba w sekwencji liniowej. W strukturach drugorzedowych
(helisa alfa, lancuch beta ) kolejne aminokwasy ulozone sa wedlug
charakterystycznego, powtarzajacego sie motywu geometrycznego oraz posiadaja
charakterystyczny i powtarzajacy sie uklad donorow i akceptorow wiazan
wodorowych.
Uwagi dotyczace
ponizszego opisu.
1. Wszystkie komendy, ktore
nalezy wpisac, podane sa w kolorze czerwonym.
2. Teksty pisane wieksza
czcionka stanowia informacje , ktore uzyskuje sie na monitorze.
URUCHOMIENIE PROGRAMU
(login, password,
desktop, unix shell)
Zamiast nazwy
programu “unix”, w zaleznosci od komputera pojawi sie jego nazwa:
leucine1%,
valine1%, glycine1%
Program 'O'
uruchomimy z valiny lub glicyny, zatem osoby pracujace na leucynie musza sie
polaczyc (np. z valina).
leucine1% telnet valine (odpowiedziec jak zwykle na pytania zadawane przez komputer)
Za pomoca komendy cd (change directory) przejdz
do swojego roboczego katalogu a nastepnie do katalogu "o", w ktorym
wykonujesz cwiczenie.
Komenda ls (list)
valine1% ls
Komenda more 9pap.pdb
pozwoli Tobie sprawdzic, czy wspolrzedne rzeczywiscie sa tam zapisane. (9pap to
kod jednej ze struktur PAPAINY znajdujacych sie w PDB).
valine2% more 9pap.pdb
Po sprawdzeniu
zawartosci zbioru uruchom program ‘O’ wpisujac komende o.
valine1% > o
W naszym oknie
(Terminal window) ukaze sie kilka informacji o programie ‘O’. Nastepnie ukaza
sie pytania:
O> Define an O file (terminate with blank): papaina.o
O> Define an O file (terminate with blank): [enter]
………………………………………………………………………….
O> Do you want to use the display [Yes]: [enter]
Obok okna
tekstowego ukazuje sie okno graficzne (graphic window), w ktorym
najprawdopodobniej nic sie nie ukazalo oprocz wiersza z roznymi opcjami u gory
okna ). Wybierz z nich opcje menus a dalej user menu. Najezdzajac mysza na kwadracik w lewym,
gornym rogu możesz uzyskac caly zestaw potrzebnych w cwiczeniu komend. Ustaw je
w takim miejscu aby było Tobie najwygodniej.
Nastepnie musisz
wczytac wspolrzedne PAPAINY i sekwencje do bazy danych programu ‘O’.
s_a_i
Sam> Name of input file : 9pap.pdb
Sam> O associate molecule name : 9pap
Ukazuja sie
informacje o liczbie atomow i reszt aminokwasowych w czasteczce.
Jak na razie
czasteczka zostala umieszczona w bazie programu 'O', teraz trzeba ja
zwizualizować.
TWORZENIE OBRAZU CZASTECZKI
PAPAINY
W oknie tekstowym
(lub graficznym) musisz zdefiniowac, o ktora czasteczke chodzi (nie zapomnij o
tym, aby kursor znajdowal sie w tym oknie, z ktorego chcesz korzystac).
mol 9pap
Komenda mol pochodzi od molecule_name,
9pap to nazwa czasteczki, jaka nadales przed chwila wczytanej strukturze.
Teraz musisz stworzyc
i nazwac obiekt:
obj nazwa
Obj od object,
zamiast “nazwa” wpisz dowolna nazwe, np. pap. Nastepnie musisz zdecydowac jak
Twoj obiekt ma wygladac. Czy ma to byc tylko lancuch glowny czasteczki bialka z
polaczonymi atomami C-alfa czy maja byc widoczne reszty aminokwasowe, czy tez
ma byc widoczny tylko fragment lancucha. W naszym wypadku chcemy pokazac caly
lancuch glowny zaznaczony atomami C-alfa
ca (enter)
mol>
ca zone [all molecule]: [enter]
Program zapytal czy
ma byc narysowany caly lancuch? Potwierdz.
Teraz musisz
zakonczyc operacje wpisujac end.
end
W oknie graficznym
w menus/object ukazala sie nazwa Twojego
obiektu z dopiskiem on czyli „wlaczony” (jeśli
jest off znaczy to, ze obiekt jest wylaczony i
wystarczy najechac na nazwe obiektu i nacisnac lewym klawiszem myszy).
Jesli miales
szczescie, to pojawil sie obraz Twojego obiektu. Jesli szczescia nie miales,
musisz sprowadzic obraz „sila” do okna graficznego. Uzywajac komendy
centrowania mozesz sprowadzic wybrany atom czasteczki na srodek okna
graficznego.
Centrowanie w
punkcie ciezkosci czasteczki.
ce_zo
9pap 1 212
Centrowanie na
dowolnym atomie (np. atom nr 10).
ce_at 9pap 10
Po wykonaniu jednej
z nich, w oknie graficznym pojawi sie czasteczka (jednokolorowa).
Mozna ja obracac,
powiekszac, pomniejszac. Osoby korzystajace z valiny moga uzywac osmiu pokretel
(sprawdzcie sami, jak dzialaja). Siedzacy przy leucynie i glicynie musza
posluzyc sie mysza:
prawy klawisz - obrot xyz
prawy klawisz + shift -
przesuniecie wzdluz x i y
prawy klawisz + shift +
srodkowy klawisz - przesuniecie wzdluz z
prawy klawisz + srodkowy
klawisz - powiekszanie/pomniejszanie
prawy klawisz + lewy
klawisz - grubosc warstwy
Można tez wybrac z menus opcje fake dials (sprawdz sam jak to dziala albo zapytaj prowadzacego).
ANALIZA STRUKTURY DRUGORZEDOWEJ
Komenda yasspa
sluzy do wyszukiwania w czasteczce fragmentow o specyficznej strukturze
drugorzedowej (helisy, lancuchy beta ).
yasspa
Util>......
Util>......
Util>......
(informacje o
liczbie reszt tworzacych helisy)
yasspa
9pap beta 0.8
Util>.....
Util>.....
(informacje o
liczbie reszt tworzacych lancuchy beta )
Komputer juz wie
ktore rejony PAPAINY to helisy, a ktore lancuchy beta . Ty nie wiesz. Co zatem zrobic,
aby sie dowiedziec? Pokoloruj helisy na czerwono, lancuchy beta na zielono, a
reszte na bialo.
Tworzymy obiekt z
zaznaczonymi na kolorowo strukturami drugorzedowymi.
mol 9pap
pai_zon ; ; white
pai_prop res_2ry
= beta green
pai_prop res_2ry
= alpha red
obj yasspa ca ;
end
W menus/object masz juz trzy obiekty. Uaktywnij tylko yasspa (klikniecie mysza na nazwy pozostalych
obiektow powoduje ich wygaszenie). Teraz znajdz najdluzsza helise mierzac
dlugosc za pomoca komendy (w menu) distance_define
przez klikniecie na nia oraz na dwa atomy, miedzy ktorymi mierzymy odleglosc.
(Aby usunac zbedne czerwone napisy z rysunku, nalezy uruchomic komende Clear_ID oraz Trig_reset).
Wybierz najdluzsza
helise i obroc nia w taki sposob, aby jej os glowna byla prostopadla do
plaszczyzny ekranu. Pamietaj, helisa powinna byc tak zorientowana, aby atom
C-alfa o nizszym numerze byl blizej Ciebie, a atom C-alfa konczacy
helise-dalej. Numery reszt sprawdzamy, jak juz na pewno zauwazyles, przez
klikniecie odpowiedniego atomu. Zauwaz, ze u gory okna graficznego ukazaly sie
jego wspolrzedne.
Kilka informacji na temat helis.
|
Typ
helisy |
Liczba
reszt aminokwasowych na zwoj |
Liczba
atomow w pierscieniu zamknietym przez wiazanie wodorowe |
Oznaczenie |
|
alfa |
3.6 |
13 |
|
|
pi |
4.4 |
16 |
|
|
|
3.0 |
10 |
Sprawdz z jaka
helisa masz do czynienia liczac ile reszt aminokwasowych (wiazan C-alfa C-alfa )
przypada na jeden skret. Wykonaj to dla kilku helis.
KATY TORSYJNE
Charakterystyczna
cecha lancuchow polipeptydowych sa katy torsyjne Phi i Psi
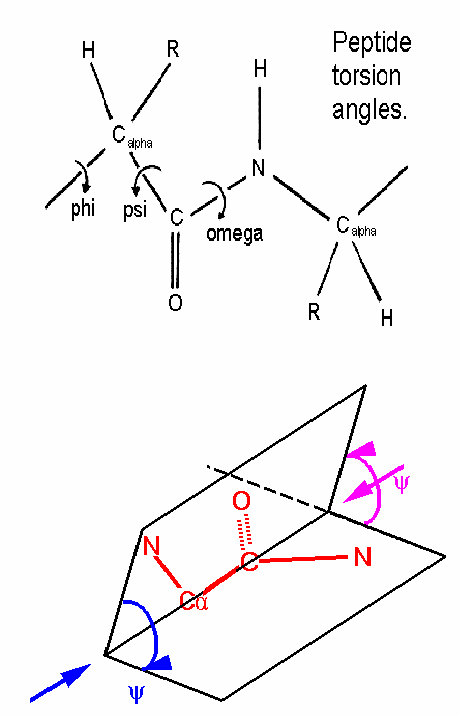
Wartosci tych katow
sa charakterystyczne dla struktur drugorzedowych i odpowiadaja uprzywilejowanym
obszarom wykresu Ramachandrana.
Indyjski naukowiec
Ramachandran obliczyl wielkosc energii czasteczki dla roznych kombinacji katow psi
i phi dla prostego dipeptydu alanyloalaniny (AA). Stwierdzil, ze tylko dla
niektorych par katow torsyjnych phi i psi z przedzialu od -180 do 180° energia
jest wystarczajaco niska, aby taka struktura mogla istniec. Pozostale
konformacje sa niedozwolone.
Zadanie polega na
zbadaniu, do jakich obszarow wykresu Ramachandrana naleza grupy peptydowe w
znalezionych przez Ciebie elementach struktury drugorzedowej PAPAINY.
Postepowanie:
Najpierw analizujesz helisy. Wybierz z
menu komende PhiPsi i kliknij na atom C-alfa w helisie. W lewym gornym
rogu (druga linia ) ukazuja sie wartosci tych katow torsyjnych. Zanotuj te
wartosci dla kilku reszt w helisie. Podobnie postepujesz z lancuchami beta.
Teraz na papierze milimetrowym narysuj uklad wspolrzednych (Phi to os pozioma,
-180 do 180; Psi to os pionowa, -180 do 180) i zaznacz w nim punkty
odpowiadajace parom katow znalezionym w strukturze.
Jakie wnioski
mozesz wyciagnac z tego wykresu co do sredniej wartosci katow torsyjnych dla
helis i arkuszy beta.
MODELOWANIE PAPAINY
Jednym
z etapow w badaniach strukturalnych makroczasteczek jest modelowanie
molekularne. Polega ono na takim manipulowaniu polozeniami lancuchow glownych i
bocznych modelu (zmiana katow torsyjnych) aby mozliwie najwierniej odtworzony
zostal rzeczywisty obraz czasteczki. Sluza do tego mapy gestosci elektronowej
liczone na podstawie czynnikow struktury naszego modelu (fc) i danych
eksperymentalnych (fo) i wyswietlane w programie 'O'. Najczesciej korzysta się
z mapy 2fo-fc (kolor niebieski) i mapy roznicowej fo-fc (kolor czerwony i
zielony). Mapa roznicowa wskazuje nam gdzie w modelu wystepuja bledy, które
nalezy poprawic. Jesli jakis lancuch boczny modelu przyjmuje inna konformacje
niż w rzeczywistej strukturze uwidoczni sie to na mapie obecnoscia mapy ujemnej
(czerwonej) tam gdzie "cos" jest a nie powinno byc; i mapy dodatniej
(zielonej) tam gdzie powinno "cos" byc a tego nie ma. Ujemna gestosc
elektronowa (czerwona) wystapi zatem w przypadku nadmiaru elektronow a dodatnia
(zielona) w przypadku niedoboru elektronow. Dwa ponizsze przyklady ilustruja to
zjawisko. Na rysunku A lancuch boczny przyjmuje zla konformacje (obecnosc
roznicowej gestosci elektronowej). Zmiana katow torsyjnych lancucha bocznego
pozwolila nadac mu prawidlowa (rzeczywista) orientacje (rysunek B).
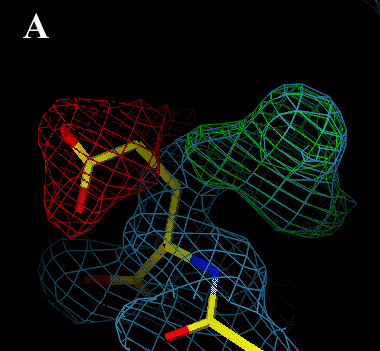
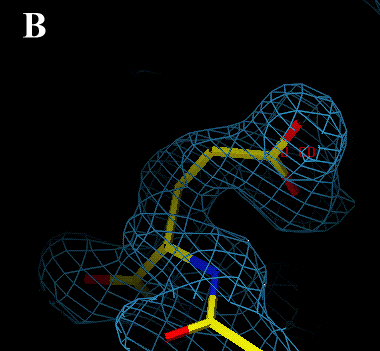
Zadanie
polega na przemodelowanie fragmentu lancucha miedzy resztami 17 i 19 oraz czasteczek
wody 301 i 302. Sluza do tego komendy z menu:
tor_res - zmieniajaca katy torsyjne lancucha glownego (phi i psi)
oraz lancucha bocznego (chi1, chi2, chi3 ...)
move_atom - umozliwiajaca przesuwanie atomu w dowolnym kierunku
(uzywana w przypadku czasteczek wody)
fm_file – sluzace do wczytywania map gestosci elektronowej 2fofc i
fofc do programu O
Pierwsza
rzecza, która nalezy zrobic jest utworzenie czasteczki z widocznymi wszystkimi
atomami. Wprawdzie wiesz juz jak to zrobic ale wole przypomniec.
mol 9pap obj nazwa zo
mol> zo zone [all molecule]: [enter]
end
Wygas
wszystkie zbedne obiekty pozostawiajac jedynie ten ostatni.
Najlatwiej
zaczac od wody. Nalezy zatem wycentrowac czasteczke na atomie tlenu wody 301.
Jak to zrobic?
ce_at nazwa 301 O1
Teraz
wczytaj mapy gestosci elektronowej. W oknie tekstowym wpisz komende:
fm_file
Fm> File name?
[]: ../../2fofc.map
Fm>
Name of this map? [Q1]: [enter]
W tym momencie pojawia sie kilka informacji na temat wczytanej mapy.
Podobnie postepujemy z nastepna mapa - fofc.map znajdujaca się w Twoim
katalogu. Zapamietujemy ja jako Q2 i Q3 (trzeba zatem wczytac ja dwa razy). Pod
opcja Density mozna teraz znalezc wczytane
mapy Q1, Q2, Q3. „Wyciagnij” je (lewy kwadracik) i ustaw parametry w
nastepujacy sposob:
Mapa Q1 level=1.2, kolor niebieski (mapa
2fofc)
Mapa Q2 level=-3.0, kolor czerwony (ujemna mapa
fofc)
Mapa Q3 level=3.0, kolor zielony (dodatnia
mapa fofc)
Za pomoca komendy move_atom przesun atom tlenu w to miejsce, ktore
wydaje sie Tobie najodpowiedniejsze (to, ktore sugeruje mapa roznicowa). Nowa
pozycje wody potwierdz komenda YES lub cofnij operacje komenda NO.
Podobnie
postepujemy w przypadku lancuchow bocznych reszt 17-19. Wycentruj najpierw na
reszcie 17, wybierz opcje tor_res i do dziela. W lewym dolnym rogu okna graficznego ukazala
sie informacja, ktore pokretlo odpowiada jakiemu katowi torsyjnemu. Uzytkownicy
leucyny i glicyny musza poradzic sobie inaczej (fake dials z opcji menus). Katow
torsyjnych lancucha glownego phi i psi lepiej nie zmieniaj. Zajmij sie tylko
lancuchem bocznym (chi1, chi2 ...) Jeśli nie wszystkimi katami mozesz ruszac
sprawdz jak dziala komenda dial_prev i dial_next. Jeśsli uznasz, ze lancuch boczny ma juz prawidlowa
orientacje potwierdz to komenda YES. Kiedy zakonczysz modelowanie wszystkich trzech reszt
zapisz zmiany komenda save.
Mostek
dwusiarczkowy, to polaczenie kowalencyjne (S-S) pomiedzy dwiema, zwykle
odleglymi w sekwencji resztami cysteinowymi (Cysteina, Cys, posiada w lancuchu
bocznym grupe tiolowa -S-H). Mostki dwusiarczkowe to jedyne przypadki
zaklocenia, poprzez rozwidlenie, absolutnie liniowej struktury kowalencyjnej,
jaka posiada lancuch bialkowy.
A teraz sprawdz,
czy wystepuja mostki dwusiarczkowe (Cys-S-S-Cys) w PAPAINIE. Narysuj obiekt s-s
z zielonymi resztami cysteiny. Po utworzeniu obiektu za pomoca komendy distance_define zmierz odleglosci miedzy
najblizszymi atomami siarki z zielonych cystein (ostatni atom w zielonym
lancuchu oznaczony SG).
mol 9pap
pai_zon ; ; white
pain_cas
Paint>.....
Paint> Property [atom_z]: residue_type
Paint> How many cases [1] :1
Paint> Enter property values [1 2 3]
: Paint> Property value 1 : Cys
Paint> Enter 1 colour name? :
Paint> Colour [white]: green
obj s-s zo ; end
Jak myslisz, czy
otrzymane odleglosci mozna uznac za mostki dwusiarczkowe? Jaka powinna byc
odleglosc miedzy atomami siarki aby uznac ja za mostek?
NA ZAKONCZENIE COS MILEGO
Uszykuj w oknie
graficznym miejsce na cos specjalnego (wygas poprzednie obrazy widoczne w oknie
graficznym). W oknie tekstowym wpisz komende sketch_auto:
sketch_auto (kilka razy enter)
Poczekaj chwile, az
ukaze sie rysunek. Zadanie dodatkowe: wyszukaj na tym wspanialym obrazku
rownolegle i antyrownolegle arkusze beta.
Jesli chcesz
zakonczyc, wpisz komende stop lub kliknij na nia
w menu. Program zapamieta automatycznie Twoje rysunki i menu.
KONIEC
Na podstawie
"O for Morons" Gerarda J. Kleywegta opracowal Robert Janowski